GBS Pipeline Tutorial - Step 2 - Trimming
Trimming
Introduction:
Now that we have completed the process of demultiplexing we can continue to move on to the next step in the GBS pipeline. The second step is trimming the demultiplexed reads. The purpose of this step is to trim adapter sequences off the ends of the reads and to remove low quality sections of data. This step ensures that the appropriate alignments are found in step 3 and for the rest of the pipeline. If you feel like you need a refresher on command line operations you can always visit the site posted in the demultiplex tutorial.
Materials:
For this step will need to do three things: reorganize our index files, reorganize the demultiplex files and create an adapter file. The only new information that we need for this step of the pipeline is what we call an adapter file. This file will contain a list of the adapter sequences that were used in the preparation of the plate(s) sent for sequencing. For each set of the R1 and R2 reads you should have a specific set of adapter sequences that were used in the lab. These can follow a variety of names such as adapters for top and bottom strands, adapter 1 and adapter 2 and so on and so forth. At this point you should already be in possession of these adapter sequences as they were required to prepare the plate(s). An example of what these adapters should look like in fasta format is shown below.
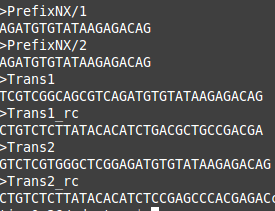
These adapters must be in fasta format for Trimmomatic to use them. The example above shows some common adapters used in sequencing. These are not going to be of much use for a real GBS run, as there are many indices used and therefore more adapters will have been designed for GBS runs. Without the adapter sequences it is difficult for trimmomatic to tell that these portions of the reads need to be removed. Failing to remove these sequences from the reads can throw the entire GBS pipeline askew. Therefore it is very important that you track down your adapter sequences and keep them on hand for this step.
Preparing the fasta file:
Once you have located your adapter sequences you must now move them into a file in fasta format. You can attempt to do this yourself but it is more than likely that someone from the bioinformatics group has already made a script to do this and may be willing to provide it to you. If you are unsure of how to do this you can contact one of the bioinformatics members to ask for help. This is the only major hurdle that is needed in preparation for running the trim step.
Re-organizing Index files and demultiplex files:
If your sequencing run has more than one lane, then we will need to perform a few more tasks before we begin. The last two things that we have to do before running the program is to do some file re-ogranizing. Remember the index files we used for each demultiplex step? We will need to combine all of those files into one large index file. You can do this however you want. You can copy them to your own local computer and modify them or you can try to learn some commands to do it through command line (it's not too difficult). This is a fairly straight forward task and can be completed fairly easily. The last thing to do is to gather all of the demultiplex files in one location. Once again, this step is necessary if you have more than one lane of sequencing runs. If you only have one lane then you can simply move forward. Otherwise you will see that there are multiple demultiplex directories if you have organized your files appropriately. The next step is to combine the demultiplex output into one directory. This can be done pretty easily by using the mv command. If you have separated your lanes into individual directories (which is the easiest) then you can start moving files around. First, create a directory where you wish to store all of the reads. This directory should be created in the same directory that you wish to continue the GBS pipeline in and should also be labelled "demultiplex" (without qutations). Then, individually go to each demultiplex output directory and use the command mv *.fastq ______ and then enter the path to the directory you have just created on the underlined space. Now rinse and repeat for all other demultiplex directories you have. Alternatively you can use the cp command instead of mv if you wish to keep the original files where they were. Now that you have all of your files organized and you have the full index file then you can start editing the GBS.conf file.
Editing the GBS.conf file:
Once again we will have to edit the "GBS.conf" file to incorporate the adapter sequence file you have acquired or created. If you still have a copy of the "GBS.conf" file on your local computer you can just modify that one and then transfer it back to the server. If you don't have it on your computer anymore you will have to go back through WinSCP and re-copy it as described in the previous demultiplexing step. Once you have the "GBS.conf" file ready to go you can open it with your choice of text editor. If you have the previous fields filled in from demultiplexing you can simply leave those as they are. The only thing you have to include is the adapter sequence file or as I will now refer to it, the trim file. Your trim file should be placed in the same directory as all of your other files. Making sure the trim file is in the right location is up to you! Don't forget to use the commands previously discussed to get files in the right locations. If you know the trim file's path relative to you, you can use that path in the GBS.conf file. That is what I have done in this situation because the trim file is located with the program (instead of with all of my GBS files). For this tutorial I am also using a trimmomatic trim file which a real GBS run should not be using. I am only using this file because I simulated my reads from a truncated E.coli genome and therefore did not perform any in-lab manipulations. For real world data you must specify the adapters that were used in the preparation of your sample which is why I will continue to emphasize it. Once you have filled in the location of the trim file to the "GBS.conf" file we are ready to begin trimming. Don't forget to set up a screen before running the trim program! It's easy to forget but things can go haywire at any point so always use a screen for data intensive processes like these. Head back to the directory that contains all of your current GBS files so far and enter the following command:
/storage/bin/Applications/GBSpipeline/GBS_pipeline.pl trim
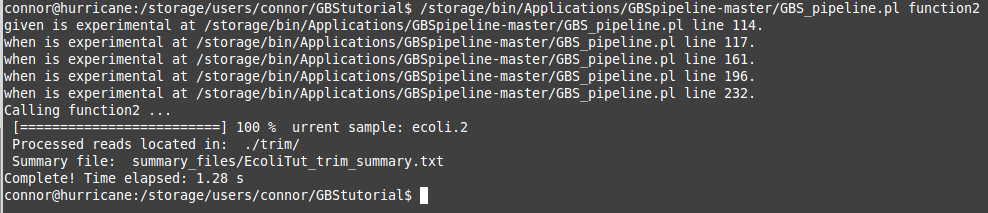
If your "GBS.conf" file was correctly filled out and your trim file was created correctly you should see some terminal output that looks like the one below!

Potential errors that may occur during this process are an improperly specified file path (a file is not where you said it would be) or an improperly formatted trim file. Usually these are simple fixes so don't be discouraged if the pipeline throws some errors at you. Once again its just part of the process! If you find that you are stuck and cannot get it to work, the bioinformatics team is availble for support. This step will also take a much longer time if your sample size is much larger. All of these programs can take days to complete! So always remember to use a screen and keep and eye on your processes using htop.
The tutorial for step 3 can be found here: http://knowpulse.usask.ca/portal/node/1772192
{kind=link}
{kind=link}