GBS Pipeline Tutorial - Step 1 - Demultiplexing
Demultiplexing
Introduction:
This is the tutorial to walk you through the first step of the GBS pipeline and to help introduce you to the command line/putty interface. If you have taken a look at the GBS flow chart I provided in the READ ME section of the tutorial then you may have already determined that this is the demultiplexing stage of the pipeline. The goal of this step is to look through all of the samples you have, remove any barcodes that are attached to the read and then to sort the files into an output directory. This tutorial aims to be as helpful as I can make it so if there are any concerns, please let one of us know!
Materials:
The materials that you will need for this step should have already been prepared in the previous tutorial. If you have not organized your files yet, I would suggest that you refer to the GBS pipeline tutorial titled: Command Line and File Organization. It provides some good knowledge on how to organize the files for a GBS run as well as some important commands for the pipeline. If you do have your files organized already then we can move on to filling the GBS.conf file!
Filling in the GBS.conf file:
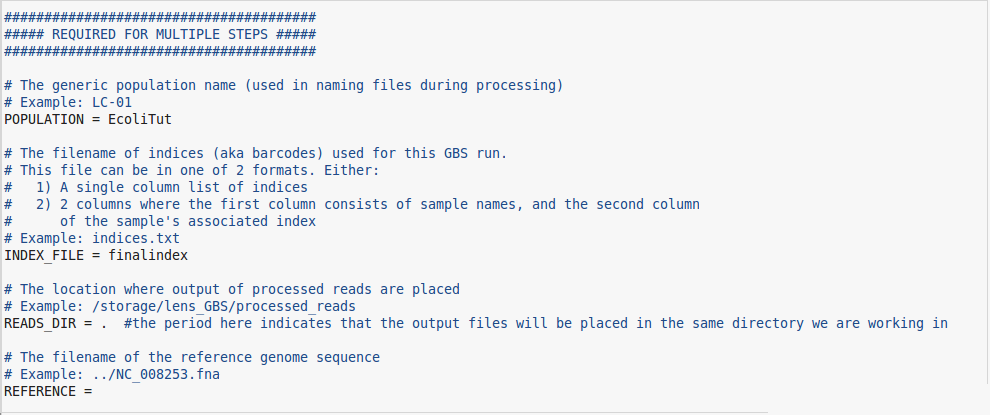
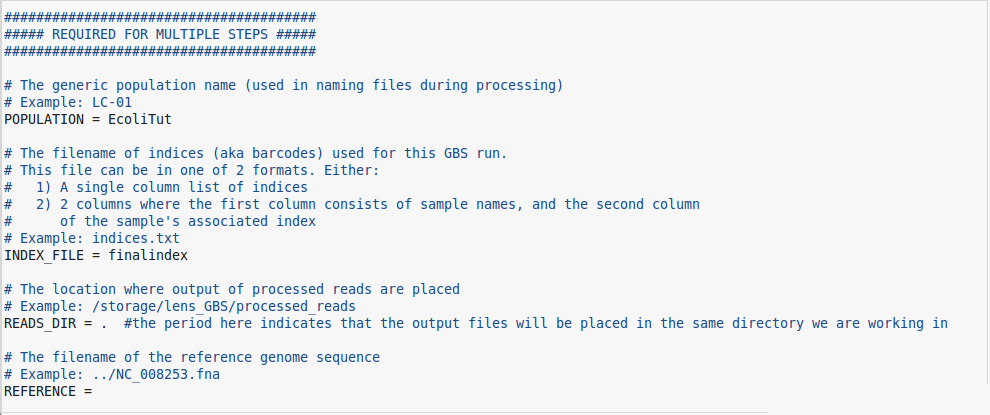
We now have the "GBS.conf" file downloaded and ready to work with. Open this file up in your text editor of choice. The next few steps may be a little bit tricky so I will add pictures with pathnames relevant to my server space. They will not be the same as yours but should be close enough for you to follow.
- The first thing you will change in the "GBS.conf" file is the POPULATION field. You can name this anything you like but again, you should use a relevant name to the experiment or project.
- The second field to fill in is the INDEX_FILE field. Here you will simply enter the name of the index file that you have downloaded from knowpulse (or created), and transferred to the server. If you have not gathered this file from the LIMS section of knowpulse then please do so. If you are unsure of where to find this file, it will be attached to the same page as the LIMS record that you have hopefully entered in knowpulse. Any other concerns about what to do can be directed to the bioinformatics group at this point.
- Moving on, in the READS_DIR field, simply place a period "." just like so but without the quotation marks. This simply tells the program that the output will be placed in the same directory you called the program from. If you want the output files to be placed somewhere else, you can specify that directory, or if you would like the pipeline to create an output directory for you, you can list the name of the directory you'd like to create here.
- The REFERENCE field can be left blank for this step. If you know the location of the reference genome, you can fill it in here, but the pipeline doesn't require it until the third step.
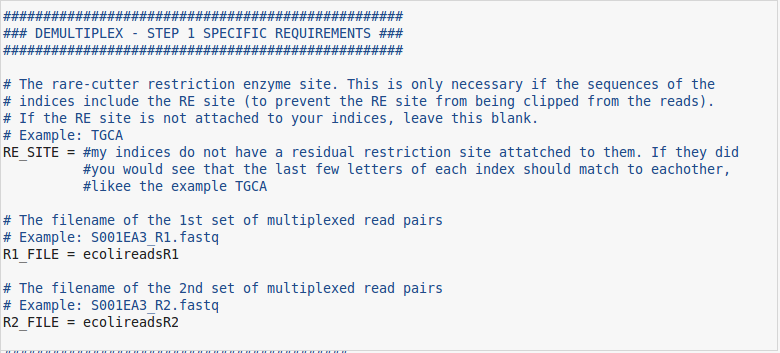
Once you have filled in the appropriate fields you can now scroll down to the DEMULTIPLEX requirements section.
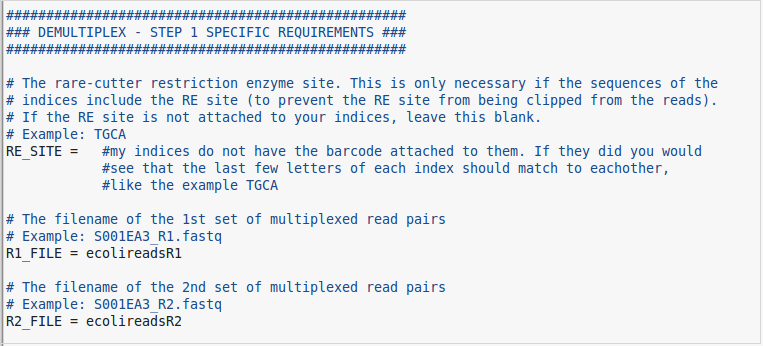
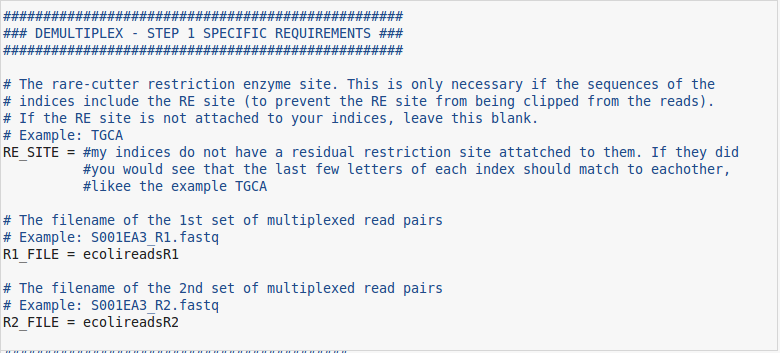
- The first field to pay attention to is the RE_SITE. Depending on how your index file is set up you may or may not need this. If part of your enzyme cut site is included in your indices, you will need to specify the part that is left over so it doesnt get cut off by the demultiplexing step. Chances are, your indices will not have a cut site present within them, so we can leave this field blank.
- The last two fields are simply the paths to the sequence files that you are working with. In my case I will be using simulated reads from an E.coli genome.
At this point, everything else can be left blank if you have filled in the appropriate fields. If you get an error when you run the first function that is okay. Errors happen all the time and are a part of working with computers. If you do get an error and are unsure of what to do then come visit the bioinformatics group. Otherwise if you have located all of the correct files and organized everything appropriately we should be ready to start the function. Below are some pictures of what my GBS config file looks like for this run. Don't forget to copy this "GBS.conf" file back into the server where all your other files are. The pipeline will need to see that it is in the same directory from where you are calling the pipeline in order to work properly.
The last thing to do before running the program is to double check that all of our files are in the right location. If you use the "ls" command you should see your sequenced reads (both R1 and R2), your index file, and the GBS.conf file that we just finished working on. So four files in total. If you do see all of these files then you are ready to run the final command! However, before you do this I would suggest that you run the command from a screen session. A screen session is almost like a separate window on the server. It allows us to log out and go about other business without needing to worry about accidentally cancelling the function. To start a new screen session, use the command:
screen -S anynameyouwant
This opens up a new screen on the server and prevents worries associated with power outages or network difficulties. To exit a screen session and return to normal server access use the keyboard command:
control+A+D
If you wish to return to a screen session that you have disconnected from you can use the command:
screen -r NameOfScreenYouWishToResume
It is possible to have multiple screen sessions open at the same time so if you ever forget which ones you have open you can use the command:
screen -r (hit the tab key twice)
This will print a list of all the screens that you currently have open on the server. If you wish to completely close a screen you should resume the screen session using the methods described above. Once you have resumed the appropriate screen, make sure that there are no processes running that are important to you. To exit a screen session while you are in it, just type exit and hit enter. However, it is good practice to cancel programs you are running before you exit the screen session (if you do not need the programs running), otherwise the system administrator will have to cancel them at a later time. To cancel a running process while in a screen use the keyboard command:
control+C
Once you have stopped any processes that are running and you are ready to close the screen, simply type the word:
exit
Then press enter and the terminal should spit you back onto the server.
If you have any questions up to this point, now is the time to ask. The bioinformatics group is available for reach from email or in person if you are on campus. This is quite a complicated process so questions are welcome! Otherwise you can run the following command below in your terminal session. Before you run the command make sure you are in the correct directory on the server (hint: use ls to see if all the files you need are present). Prior to running this command, please follow this link to learn a little more about server usage: http://knowpulse.usask.ca/portal/node/1772323.This link will take you to a quick tutorial where I discuss how to check server usage with the program htop. If you don't have time to learn about this please contact Carolyn or Larissa to have them check the server usage for you. If you are not in the right directory when the command is entered, you will most likely get an error saying that one or more files could not be found. Make sure you are in the right directory! To start the demultiplexing program, enter the command below:
/storage/bin/Applications/GBSpipeline/GBS_pipeline.pl demultiplex
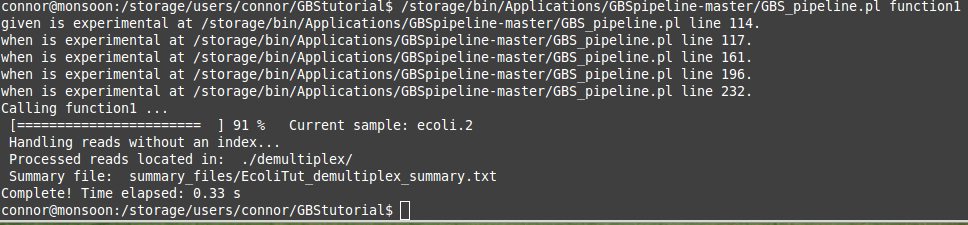
The command above can be copied and pasted into your command line and as long as you are in the correct directory on the server you should see your window spit out some text out and a progress bar will show up. Depending on your file sizes this step can take a while. The "when is experimental..... " text at the start of the run can be ignored. It should look a little bit like the picture below.
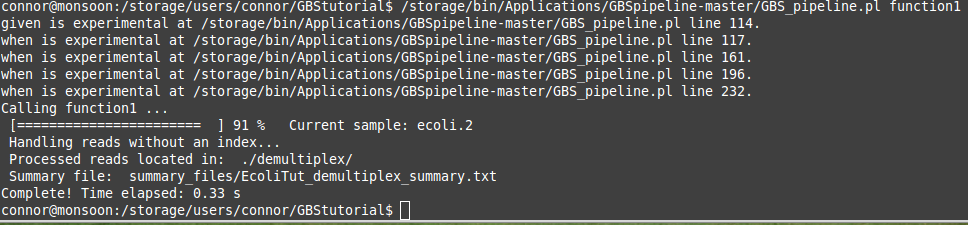
If your files are large this will take a while, as mentioned before. This is why I recommend that a screen be used to ensure that the program still runs even if your computer's network connection is dropped or the power goes out. To see if your process is still running you can open up a second terminal or putty session, log in to the server, and then type the command "htop". This will print a list of all the processes being run on the server in real-time. Your process should be one these! Sometimes they can be hard to find if the server is quite busy. Alternatively you can use the htop -u <yourusername> command. This simply filters through all of the processes and only lists those for a specific username. This step is not necessary but can be helpful if you are unsure about whether your program is running or not.
Once your program has run to completion you should have some new directories and files in your working directory. This is a good sign! These materials will be used for the next steps in the pipeline. You should see three new directories labelled summary_files, demultiplex, and tmp. It is always good practice to look over the summary files after the program has finished running. If you are not sure how to do this or are not sure what the data implies, you can come and ask us once again! We are here to help. Otherwise you should be set to move on to the next step in the GBS pipeline process which is the trimming step!
The tutorial for the trimming step can be found here: http://knowpulse.usask.ca/portal/node/1772191
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}